| Trial | Drug | Country | n | Result |
|---|---|---|---|---|
| JIKI (Sissoko et al. 2016) | Favipiravir | Guinea | 111 | Inconclusive |
| RAPIDE-BCV (Dunning et al. 2016) | Brincidofovir | Liberia | 4 | Terminated early |
| RAPIDE-TKM (Dunning et al. 2016) | TKM-130803 | Sierra Leone | 14 (12 evaluable) | Futility reached |
Testing Ebola Drugs in an Epidemic
How a decade of Ebola research produced licensed treatments — and what happens next
This is the first of two notes on Ebola trial methodology, and a companion to the Experimental Designs chapter of Global Health Research in Practice. This note follows the therapeutics arc; the companion note follows the vaccines.
The Ebola outbreak spreading through northeastern DRC and into Uganda is caused by Bundibugyo ebolavirus, the most recently discovered of the four Ebola species known to infect people. Only two previous Bundibugyo outbreaks have been recorded: one in Uganda in 2007–2008 and one in DRC in 2012. The WHO has declared a public health emergency of international concern. As of 26 May, the DRC Ministry of Health reported 105 confirmed cases (10 deaths) and 906 suspected cases (223 deaths) across Ituri, North Kivu, and South Kivu provinces; Uganda has reported seven confirmed cases (one death), several linked to travel from DRC.
Everything currently licensed for Ebola targets Zaire ebolavirus, but the licensed tools sit on two separate research tracks. The vaccines, Merck’s Ervebo and Janssen’s two-dose Zabdeno/Mvabea regimen, came from one set of programs. The therapeutics strongly recommended by the WHO, monoclonal antibodies mAb114 and REGN-EB3, came from another. None of it applies to Bundibugyo.
This note follows the therapeutics track: how researchers between 2014 and 2019 figured out how to test antiviral drugs and monoclonal antibodies during an outbreak, what came out of that work, and what it can and cannot tell us about an outbreak the licensed evidence does not cover. The vaccine story is the companion note.
How Should You Test Drugs in a Catastrophe?
The West African Ebola epidemic of 2014–16 killed more than 11,000 people. It also forced a question that researchers had debated largely in the abstract: what kind of clinical trial can you run in the middle of a collapsing health system?
Two positions emerged in late 2014.
Steven Joffe, writing in JAMA, argued for randomization from the start. The experimental agents, ZMapp foremost among them, had no demonstrated benefit in humans. Equipoise existed. Without a concurrent randomized control group, patients who receive the drug “will differ systematically from the untreated individuals with whom they are compared.” Single-arm designs couldn’t distinguish a drug that works from one given to patients who were going to survive anyway.
Adebamowo and colleagues replied in the Lancet the following month. They agreed that RCTs produce the best evidence. They disagreed that one was feasible here. Their objections were both ethical and practical. When conventional care carries a case-fatality rate approaching 70%, they argued, equipoise is hard to sustain: randomizing patients to standard care alone is hard to defend when the experimental arm holds out at least the possibility of benefit. And the practical barriers were real. Health systems were collapsing. Communities distrusted treatment centers. Families presented together, making individual randomization socially untenable. Insisting on an RCT, they wrote, “could even worsen the epidemic, by undermining trust in the Ebola treatment centres that are central to containing it.”
Adebamowo’s group proposed an alternative: run single-arm studies in parallel at different sites, designed adaptively so you could triage drugs. Roll out the ones that clearly work, discard the ones that clearly don’t, and randomize only for the uncertain middle.
Cooper and colleagues (2015) formalized this intuition into a testable design. Using simulation, they compared three designs: a conventional RCT with a fixed sample size, a sequential RCT with interim analyses, and a multi-stage approach (MSA) that began with a single-arm phase II as a triage filter. The MSA started by giving every patient the experimental treatment and monitoring outcomes against a historical benchmark. If the drug was clearly effective (survival around 80%), it could be rolled out immediately with a confirmatory study. If clearly ineffective (survival around 50%, no better than standard care), it would be rejected. For the ambiguous middle, the design escalated to a randomized comparison.
Cooper’s simulations showed the MSA could reject ineffective drugs faster than either randomized design, with fewer in-study deaths. For a field that expected to be testing multiple candidates against an unpredictable epidemic curve, speed mattered. The MSA became the design logic that every single-arm Ebola trial would follow.
The Triage Phase
Three trials used the MSA during the 2014–15 epidemic. Each was designed to do the same thing: quickly determine whether a candidate drug was worth pursuing further. None were designed to estimate efficacy.
JIKI was the largest. Sissoko and colleagues enrolled 126 patients across four treatment centers in Guinea, with 111 in the primary analysis. They gave six reasons for not randomizing, from community distrust to the ethical difficulty of randomizing family members who presented together. The clinical result was inconclusive; the confidence intervals for mortality in every stratum included the target values. But JIKI produced something more durable than a drug verdict. Mid-trial, the investigators discovered that baseline RT-PCR cycle threshold (Ct) value was the dominant prognostic factor: 20% mortality among patients with Ct ≥ 20 versus 91% among those with Ct < 20. That discovery, shared at a conference in early 2015, was carried into every subsequent major Ebola trial.
The two RAPIDE trials showed how fragile outbreak research is in practice. RAPIDE-BCV tested brincidofovir in Liberia and enrolled four patients before the manufacturer withdrew and the epidemic waned. RAPIDE-TKM tested TKM-130803 in Sierra Leone; the futility boundary was crossed after 14 patients had received the drug, with day-14 survival of 27% in 12 evaluable patients against a target of 55%. The RAPIDE-TKM investigators described their purpose explicitly: a “single-arm trial to generate early evidence of the effectiveness of TKM-130803.”
The MSA framework did what it was designed to do. It cleared three drugs from the field quickly, with minimal patient exposure to treatments that weren’t working. But the three candidates it cleared had entered the epidemic with little supporting human or animal data. ZMapp was a different case.
The First Randomized Trial
ZMapp, a triple monoclonal antibody cocktail produced in tobacco plants, had supporting nonhuman primate data the MSA candidates didn’t, with strong protection in macaques when treatment began within five days of inoculation. It was also the agent at the center of the 2014 debate, the drug Joffe specifically cited when arguing for randomization. With that level of preclinical evidence behind it, ZMapp was the candidate a US/Liberia consortium — the Partnership for Research on Ebola Virus in Liberia, or PREVAIL — chose to test with randomization from the start, bypassing the triage step.
PREVAIL II ran from March to November 2015, in parallel with the MSA single-arm trials rather than after them. It randomized 72 patients to standard care alone or standard care plus ZMapp, across sites in Liberia, Sierra Leone, Guinea, and the United States. Randomization was stratified by baseline Ct value (JIKI’s contribution, already absorbed into the design) and by country.
The clinical result was suggestive but inconclusive. Twenty-eight-day mortality was 22% in the ZMapp group versus 37% in controls. The Bayesian posterior probability of ZMapp’s superiority reached 91.2%, short of the prespecified 97.5% threshold. The 95% credible interval for the absolute mortality difference ran from −34 to +6 percentage points. The trial had been designed for 200 patients, but enrollment ended as eligible new cases declined and the outbreak was declared over. The epidemic ended before the trial could.
The drug result was ambiguous, but the methodological one was not. PREVAIL II demonstrated something the 2014 debate had treated as an open question. The authors stated it plainly: “the trial did succeed in establishing that it is indeed feasible to conduct a randomized, controlled trial in the context of a major public health emergency despite the challenges involved.” The practical objections Adebamowo raised—community distrust, logistical chaos, collapsing systems—had been real, but turned out not to be insurmountable.
PREVAIL II also created a practical bridge to the next trial. ZMapp’s 91.2% posterior probability was too uncertain to declare it effective, but too suggestive to ignore. It became the natural active comparator for the next generation of candidates: not a placebo, but a drug with a plausible but unproven benefit against which newer therapies could compete.
The Platform Trial
Three years later, the next Ebola outbreak struck eastern DRC, this time in an active conflict zone. The PALM trial (Pamoja Tulinde Maisha, “Together Save Lives”) put the previous five years of design work to use in a single trial.
PALM randomized 681 patients across four treatment centers to one of four arms: ZMapp (the active control), remdesivir, mAb114, or REGN-EB3. The allocation was 1:1:1:1, stratified by Ct value (JIKI’s discovery, carried forward once more).1
The design solved problems its predecessors hadn’t. It was multi-arm: four candidates evaluated simultaneously rather than one at a time. It was adaptive: pre-specified interim analyses could drop arms that weren’t working. And it ran in conditions that would have seemed impossible to anyone following the 2014 debate. Violence at two participating centers forced temporary evacuations. The work resumed each time, and the trial infrastructure held.
At an interim analysis in August 2019, the data safety monitoring board recommended stopping the ZMapp and remdesivir arms. Twenty-eight-day mortality: ZMapp 49.7%, remdesivir 53.1%, mAb114 35.1%, REGN-EB3 33.5%. mAb114 and REGN-EB3 were superior (P = 0.007 and P = 0.002, respectively, versus ZMapp).
Show code
palm <- tribble(
~arm, ~population, ~deaths, ~n,
"ZMapp", "Overall", 84, 169,
"Remdesivir", "Overall", 93, 175,
"mAb114", "Overall", 61, 174,
"REGN-EB3", "Overall", 52, 155,
"ZMapp", "High viral load (Ct ≤22)", 60, 71,
"Remdesivir", "High viral load (Ct ≤22)", 64, 75,
"mAb114", "High viral load (Ct ≤22)", 51, 73,
"REGN-EB3", "High viral load (Ct ≤22)", 42, 66,
"ZMapp", "Low viral load (Ct >22)", 24, 98,
"Remdesivir", "Low viral load (Ct >22)", 29, 100,
"mAb114", "Low viral load (Ct >22)", 10, 101,
"REGN-EB3", "Low viral load (Ct >22)", 10, 89
) |>
mutate(
mortality = 100 * deaths / n,
arm = factor(arm, levels = c("ZMapp", "Remdesivir", "mAb114", "REGN-EB3")),
population = factor(population,
levels = c("Overall",
"High viral load (Ct ≤22)",
"Low viral load (Ct >22)")),
continued = arm %in% c("mAb114", "REGN-EB3")
)
ggplot(palm, aes(x = arm, y = mortality, fill = continued)) +
geom_col(width = 0.7) +
geom_text(aes(label = sprintf("%.1f%%", mortality)),
vjust = -0.5, size = 3.4, color = "grey25") +
facet_wrap(~ population) +
scale_fill_manual(values = c(`TRUE` = ghr_blue, `FALSE` = ghr_slate),
guide = "none") +
scale_y_continuous(limits = c(0, 100), expand = expansion(mult = c(0, 0.05))) +
labs(x = NULL, y = "28-day mortality (%)") +
theme_minimal(base_size = 13) +
theme(panel.grid.major.x = element_blank(),
axis.text.x = element_text(angle = 30, hjust = 1),
strip.text = element_text(face = "bold"))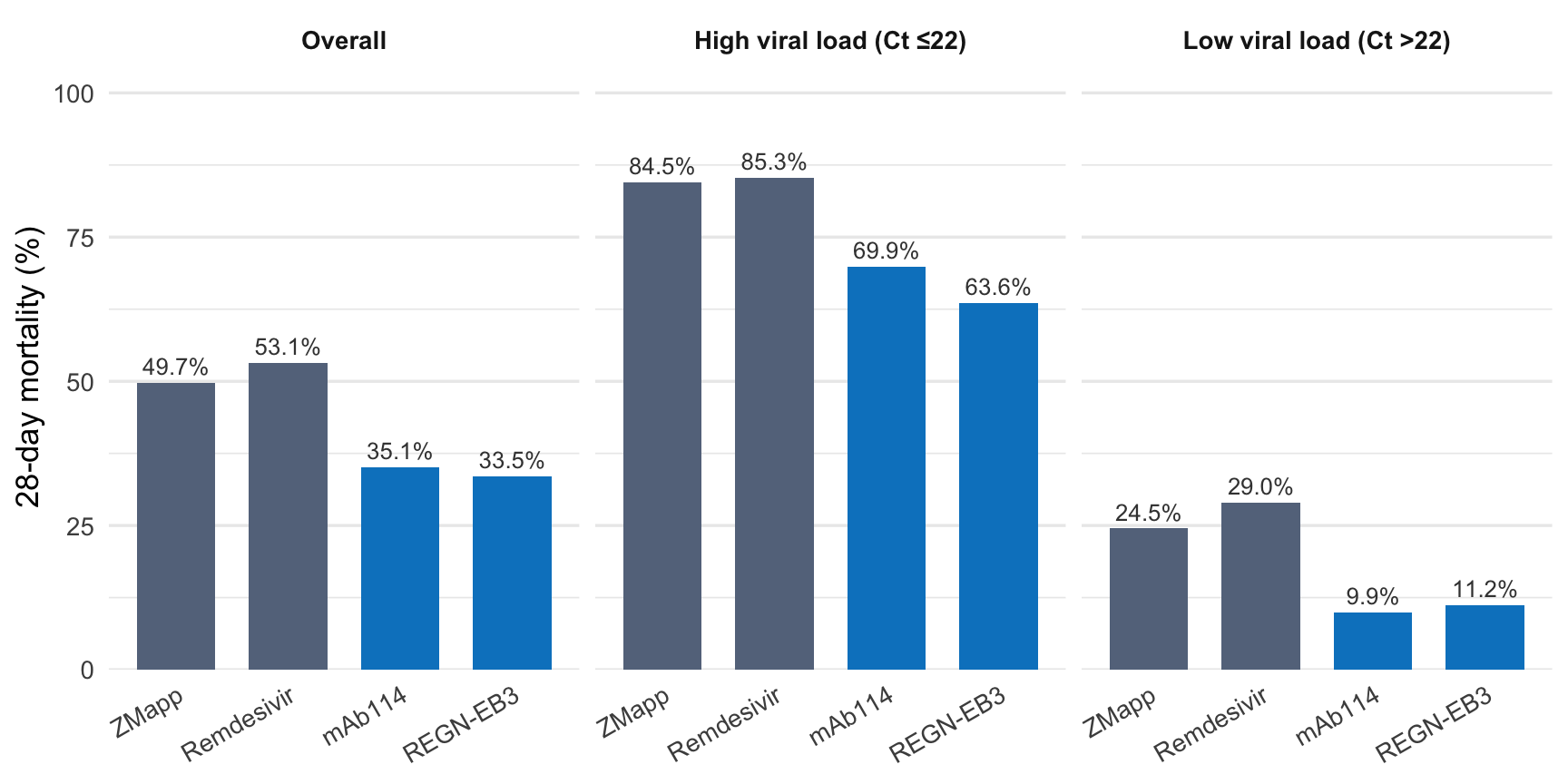
PALM was the methodological capstone. In five years, the field went from debating whether you could randomize during an outbreak to running a four-arm adaptive platform trial in an active war zone. Each step was shaped by what the previous step couldn’t do. The single-arm trials cleared the field but couldn’t estimate efficacy. PREVAIL II proved randomization was feasible but was underpowered. PALM combined the lessons: multiple arms for efficiency, adaptive stopping for ethics and speed, Ct stratification for precision, and an active comparator instead of a placebo because PREVAIL II had made pure equipoise against standard care harder to defend.
From Trial to Guideline
The WHO 2022 therapeutics guideline translated PALM’s findings into clinical policy. The underlying evidence synthesis, a systematic review and network meta-analysis by Gao and colleagues, screened 7,840 records and found exactly two RCTs meeting inclusion criteria: PREVAIL II and PALM, 753 patients combined. On that basis, the Guideline Development Group issued three recommendations:
- Strong recommendation for treatment with mAb114 or REGN-EB3 for patients with RT-PCR confirmed EVD
- Conditional recommendation against remdesivir
- Conditional recommendation against ZMapp
A decade of work produced two randomized trials, 753 patients, and one strong recommendation. Outbreaks are rare, unpredictable, and operationally hostile to research, and two RCTs is a remarkable output for a disease whose outbreaks are measured in months.
Does the Field Start Over?
The 2026 Bundibugyo outbreak finds the therapeutics field in a familiar but stronger position than a decade ago.
Familiar, because there is again no proven treatment. The clinical evidence from the last decade is Zaire-specific, and it does not transfer. mAb114 and REGN-EB3 target the Zaire glycoprotein, and antibodies like these are largely species-specific. The antibodies known to neutralize Bundibugyo are different molecules entirely, isolated from Bundibugyo survivors. There is little reason to expect the licensed drugs to work against this species, and no trial has tested them. The WHO recommendations, built on PALM, do not extend past Zaire ebolavirus.
Stronger, because the methodological lessons do transfer, even when the drugs don’t. The field now knows how to triage candidates quickly, how to randomize ethically under outbreak conditions, and how to run an adaptive platform trial in a conflict zone. None of that has to be reinvented.
What doesn’t yet exist is the infrastructure to put that know-how to work the moment it’s needed: multi-outbreak registries that pool data from small, scattered events; preregistered dormant protocols that activate when a non-Zaire outbreak is confirmed; therapeutic candidates designed against features conserved across the genus rather than against one species’s glycoprotein. The know-how is in hand; the standing capacity is not. Building it requires investment between outbreaks, when urgency is lowest and funding is probably hardest to find.
Take-Home Messages
The path to licensed Ebola treatments took a decade and was shaped by an explicit methodological debate. The progression from single-arm triage designs to a multi-arm platform RCT was deliberate, with each step responding to what the previous one couldn’t do.
Single-arm triage designs cleared the field efficiently but couldn’t estimate efficacy. The MSA framework rejected ineffective drugs quickly. Proving a drug works required randomization, which PREVAIL II showed was feasible and PALM delivered at scale.
A discovery from one trial reshaped the trials that followed. JIKI’s mid-trial finding that Ct value dominates prognosis was carried into every subsequent major Ebola trial, including PALM. Sometimes a trial’s most important contribution has nothing to do with the drug it tested.
The strongest evidence is species-specific. WHO’s 2022 strong recommendation for mAb114 and REGN-EB3 covers Zaire ebolavirus only. Whether those tools retain useful activity against Bundibugyo is an open question.
Footnotes
The primary analysis used a modified Boschloo’s exact test. Response-adaptive randomization was not used; allocation remained 1:1:1:1 throughout. The full statistical analysis plan and adaptive monitoring rules are described in the supplementary appendix to Mulangu et al. 2019.↩︎