How the same crisis produced two licensed vaccines — by two very different standards of evidence
Published
May 28, 2026
This is the second of two notes on Ebola trial methodology, and a companion to the Experimental Designs chapter of Global Health Research in Practice. The first note followed the therapeutics arc; this one follows the vaccines.
In this episode of Research Notes, I talk with Dr. Ana Maria Henao-Restrepo, former WHO scientist and one of the leaders of the groundbreaking Ebola ring vaccination trial conducted during the 2014–2015 West Africa outbreak. The trial ultimately demonstrated that the rVSV-ZEBOV vaccine was highly effective and helped establish a new model for evaluating vaccines during public health emergencies.
We find ourselves once again confronting an Ebola outbreak—this time the Bundibugyo species—without an approved vaccine against it. As of the WHO’s PHEIC determination on 17 May 2026, eight laboratory-confirmed cases and 246 suspected cases (with 80 suspected deaths) had been reported in Ituri Province, with suspected cases spread across Ituri and North Kivu and travel-linked cases reported in Kampala, Uganda.
The last time Ebola vaccines were tested in an outbreak, the result was two licensed products against a different species, Zaire ebolavirus. These vaccines reached licensure by very different routes: one proven effective in people, the other only in animals.
Merck’s Ervebo (rVSV-ZEBOV)1 is a single-dose replicating-vector vaccine. It was tested in humans through a novel outbreak-adapted ring trial.
Janssen’s Zabdeno/Mvabea is a two-dose regimen (Ad26.ZEBOV prime and MVA-BN-Filo boost) given roughly eight weeks apart. Its efficacy was never tested in humans. The regimen takes roughly two months to take full effect, so it couldn’t be evaluated against an exposure that follows transmission chains in real time. Instead, its protective effect was inferred from animals.
Show code
tribble(~Dimension, ~`Ervebo (Merck)`, ~`Zabdeno/Mvabea (Janssen)`,"Platform", "rVSV-ZEBOV, single-dose replicating vector", "Ad26.ZEBOV + MVA-BN-Filo, 2-dose prime-boost","Doses / timing", "1 dose", "2 doses, ~8 weeks apart","Onset of protection", "Rapid (single dose; usable in active outbreaks)", "~8 weeks after second dose (prophylactic use only)","Basis for licensure", "Human efficacy (ring RCT)", "Immunogenicity + immunobridging from animals","Human efficacy measured?", "Yes (100%, ring trial)", "No — inferred from animal challenge","EC authorization", "11 Nov 2019 (conditional)", "1 Jul 2020 (exceptional circumstances); withdrawn 1 May 2026 at MAH request") %>%kable(align =c("l", "l", "l")) %>%kable_styling(bootstrap_options =c("striped", "hover"), full_width =FALSE)
Table 1: Two Zaire ebolavirus vaccines, two evidentiary standards. Ervebo’s efficacy was measured in humans; the Janssen regimen’s was inferred from animals via immunobridging.
Dimension
Ervebo (Merck)
Zabdeno/Mvabea (Janssen)
Platform
rVSV-ZEBOV, single-dose replicating vector
Ad26.ZEBOV + MVA-BN-Filo, 2-dose prime-boost
Doses / timing
1 dose
2 doses, ~8 weeks apart
Onset of protection
Rapid (single dose; usable in active outbreaks)
~8 weeks after second dose (prophylactic use only)
Basis for licensure
Human efficacy (ring RCT)
Immunogenicity + immunobridging from animals
Human efficacy measured?
Yes (100%, ring trial)
No — inferred from animal challenge
EC authorization
11 Nov 2019 (conditional)
1 Jul 2020 (exceptional circumstances); withdrawn 1 May 2026 at MAH request
The Ervebo Ring Trial
The vaccine candidate rVSV-ZEBOV entered the West African epidemic with strong protection in nonhuman primates and no efficacy data in humans. The preclinical record summarized in the final-results trial paper is striking: a single dose of the vaccine completely protected non-human primates against high-dose challenge when administered between 7 and 31 days before exposure.
Promising as that animal data was, it was not evidence that the vaccine protected people, and demonstrating efficacy in humans is far harder. No trial would vaccinate people and then deliberately expose them to Ebola. That would be a challenge trial, and while scientists run human challenge trials for some pathogens—influenza, malaria, common-cold coronaviruses—they do not for a virus that kills a large portion of people it infects.
Instead, you give a vaccine to healthy people and then wait to see what happens when some of them happen to be exposed. During a waning epidemic, incidence falls just as you are ready to measure it. If you’re conducting a conventional individually-randomized, placebo-controlled vaccine trial, you might run out of cases before you can detect an effect. And as with therapeutic drug trials, randomizing people to placebo against a plausibly protective vaccine, in the middle of a lethal outbreak, has been the subject of ethical debates.
During the West Africa outbreak, researchers borrowed a solution from the endgame of smallpox eradication: ring vaccination. Rather than vaccinate a population, they vaccinated the contacts—and the contacts of contacts—of each newly confirmed case, the ring of people most likely to be exposed next.
This ring vaccination trial was called Ebola ça Suffit! (“Ebola, that’s enough”). It had three important methodological features.
First, the cluster, not the individual, was the unit of randomization. Each ring around a confirmed case was randomized as a whole. As discussed in the Experimental Designs chapter, randomizing clusters incurs an efficiency loss relative to individual randomization. The effective sample size is smaller than the actual number of individuals.
Second, the contrast was immediate versus delayed vaccination, not vaccine versus placebo. Every ring received the vaccine; randomization determined only when: immediately or after a 21-day delay. No one was randomized to nothing. This was the ethical move that made the trial defensible in the setting. It also reframed the estimand: the trial measured the effect of immediate protection against a 21-day-delayed comparator, not against an unvaccinated control.
Third, the design was adaptive, with an α-spending strategy for interim analyses. It was built to end early if the signal was strong.
Show code
tribble(~Feature, ~Detail,"Design", "Open-label, cluster-randomized ring vaccination trial (Phase 3)","Unit of randomization", "The ring (contacts + contacts of contacts of a confirmed case)","Randomized contrast", "Immediate vs. 21-day-delayed vaccination","Vaccine / dose", "rVSV-ZEBOV, single IM dose, 2×10^7 pfu","Stratification", "Location (urban vs. rural) and ring size (≤20 vs. >20)","Primary outcome", "Lab-confirmed EVD with onset ≥10 days after randomization","Trial registration", "PACTR201503001057193") %>%kable(align =c("l", "l")) %>%kable_styling(bootstrap_options =c("striped", "hover"), full_width =FALSE)
Table 2: The Guinea ring vaccination trial (Ebola ça Suffit!). The randomized contrast was timing of vaccination, not vaccine versus placebo.
Feature
Detail
Design
Open-label, cluster-randomized ring vaccination trial (Phase 3)
Unit of randomization
The ring (contacts + contacts of contacts of a confirmed case)
Randomized contrast
Immediate vs. 21-day-delayed vaccination
Vaccine / dose
rVSV-ZEBOV, single IM dose, 2×10^7 pfu
Stratification
Location (urban vs. rural) and ring size (≤20 vs. >20)
Primary outcome
Lab-confirmed EVD with onset ≥10 days after randomization
Trial registration
PACTR201503001057193
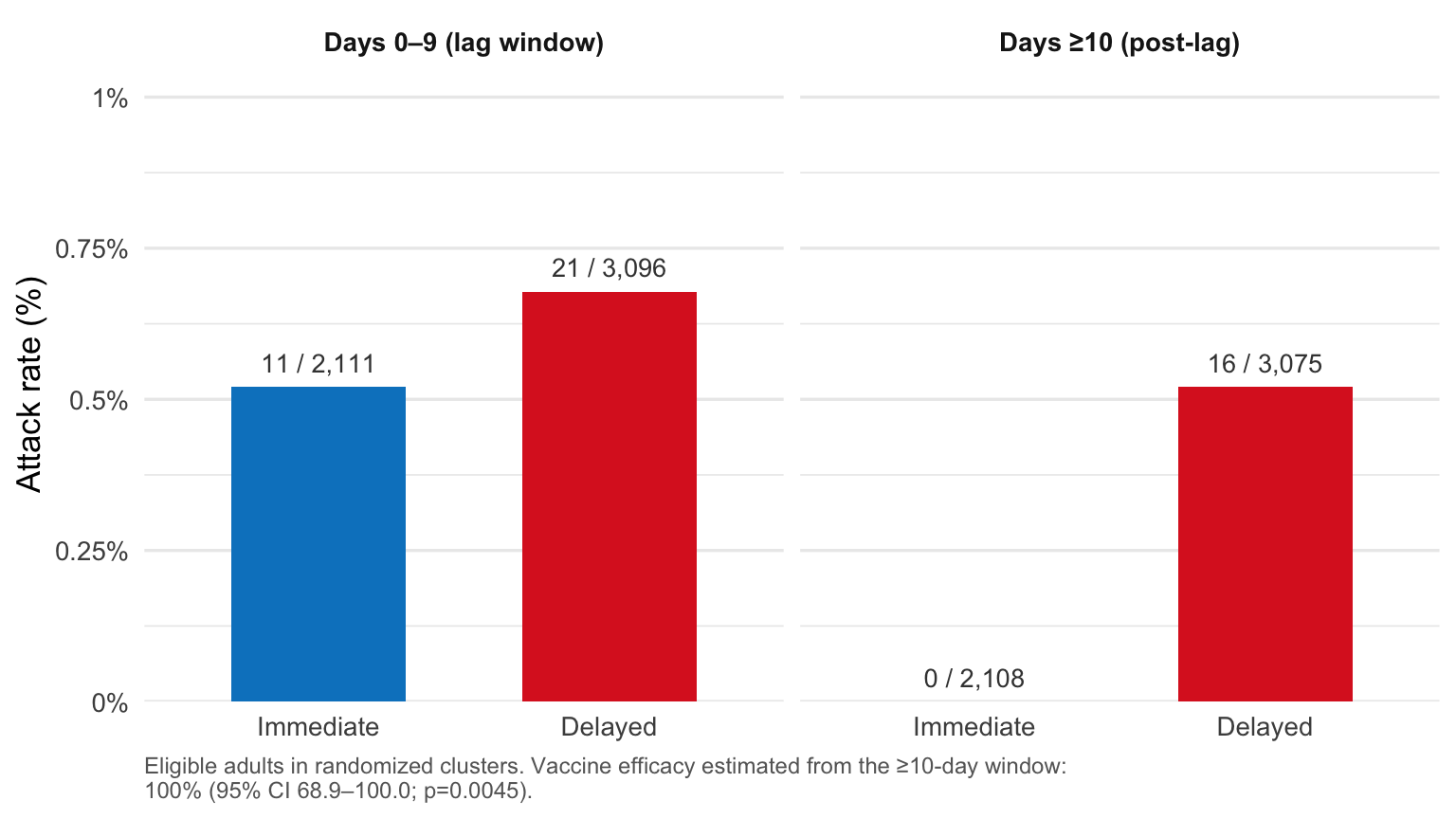
The design delivered a clear answer. No cases of laboratory-confirmed Ebola occurred 10 or more days after randomization in the immediately-vaccinated group, against 16 cases across 7 clusters in the delayed group. Vaccine efficacy was 100% (95% CI 68.9–100.0; p=0.0045).2
Figure 1: Attack rates of laboratory-confirmed Ebola virus disease in the randomized portion of the Ebola ça Suffit! trial, split by onset window. In the 10-day lag window after randomization, eligible adults in immediate and delayed clusters developed disease at similar rates (0.5% vs 0.7%). After day 10, the rates diverged sharply: 0 of 2,108 immediately-vaccinated eligible adults developed Ebola, versus 16 of 3,075 in delayed clusters. Data: Henao-Restrepo et al. (Lancet 2017), Table 4.
This promising result came with a wrinkle in how it was reached. Like most randomized trials, Ebola ça Suffit! was overseen by an independent Data and Safety Monitoring Board (DSMB), a committee that periodically reviews the accumulating data and can recommend stopping early if the evidence is decisive enough. To keep these repeated interim looks from manufacturing false positives, the trial prespecifies how strong the evidence must be to stop at each look: the α-spending boundary. At the July 2015 interim analysis, the DSMB recommended ending randomization, even though the interim result had not formally crossed that boundary.
The interim p-value was 0.0036, a bit higher than the O’Brien-Fleming success criterion of 0.0027 the authors had set for that look. The DSMB’s decision took into account the 100% point estimate together with the practical recognition that, with cases falling and the outbreak waning, the trial was unlikely to recruit many more rings. Rather than end the trial, the board recommended continuing with immediate vaccination of all new clusters, which is why the final report includes both the randomized contrast and the larger all-cluster analysis.
That is a defensible judgment under crisis conditions. The effect was large, biologically plausible given the preclinical record, and consistent with the design’s adoption in subsequent outbreaks. The evidence was strong enough to act on, and regulators moved on it with unusual speed. The European Commission granted conditional marketing authorization on 11 November 2019; WHO prequalified it within roughly 48 hours, the fastest vaccine prequalification in WHO’s history; and the US FDA approved it in December 2019.
NoteFor the careful reader: published methodological critiques
The trial was open-label. Neither participants nor field teams were masked to immediate-versus-delayed assignment. In a trial where case ascertainment depends on surveillance and reporting, lack of masking opens the door to performance and detection bias. Some worried that immediate and delayed rings were not surveilled or exposed identically.
Critics raised that bias concern in print, and the authors answered it.Metzger and Vivas-Martínez pressed on a specific mechanism: per the trial protocol, the medical study team stayed in immediate-vaccination communities to follow vaccinated participants for adverse events, while in delayed clusters the team only visited on day 0 and day 21. Differential medical-team presence for those three weeks, the critics argued, could have changed knowledge and behavior in immediate rings (awareness of symptoms, distancing, hygiene) in ways that would suppress transmission independent of the vaccine. With Ebola’s low basic reproduction number, behavioral change is a plausible alternative; in the extreme, vaccine efficacy could in principle have been as low as 0%. The authors’ reply distinguished trial-specific safety follow-up (which differed by arm) from contact tracing and risk messaging (carried out independently by national surveillance teams with identical procedures in both arms), and pointed to data inconsistent with a pure behavioral explanation: eight unvaccinated contacts in four immediate rings developed laboratory-confirmed EVD 10 or more days after randomization, and pre-vaccination case counts were similar between arms (20 immediate, 21 delayed).
The asymmetric primary-analysis population drew published critique.Zhang, Feng, and Cowling read the interim results as departing from the prespecified plan in the BMJ methods paper: they argued the prespecified primary analysis would have been a comparison of the 2,014 individuals randomly assigned to immediate vaccination with the 1,498 individuals randomly assigned to delayed vaccination, while the published analysis instead compared 2,014 individuals who actually received immediate vaccination with 2,380 eligible for delayed vaccination regardless of whether they received it. The non-symmetric handling of who counts in each arm is the substantive criticism.
The authors’ reply defended the asymmetric comparison as the correct estimand for vaccine efficacy: the hazard ratio of laboratory-confirmed EVD between eligible-and-vaccinated individuals in immediate clusters and eligible individuals in delayed clusters, with the analysis window shifted to allow for immunity development and incubation. This was distinct from effectiveness, which would include unvaccinated members of immediate rings. Read literally, the BMJ methods paper does define the primary analysis as the authors describe it (eligible-and-vaccinated immediate versus eligible delayed), making the dispute less a matter of a changed analysis plan than a disagreement about which estimand a ring vaccination trial should prioritize and whether asymmetric inclusion rules across arms can be made transparent in advance.]
Table 3: The asymmetric primary-analysis dispute, summarized. Zhang/Feng/Cowling read the prespecified plan as comparing all randomized individuals; the authors defended the asymmetric efficacy estimand they had specified and reported.
Authors — asymmetric (efficacy estimand, as published)
2,014 vaccinated-immediate
2,380 eligible-delayed
Janssen’s Regimen: Efficacy Inferred from Animals
Janssen’s regimen reached licensure by a fundamentally different route than Ervebo, one that never measured whether it protects humans.
Janssen’s regimen is a two-dose heterologous prime-boost: Zabdeno (Ad26.ZEBOV) first, then Mvabea (MVA-BN-Filo) roughly eight weeks later.3
The crucial fact for a methods reader is why it could not be licensed the way Ervebo was. A two-dose regimen given eight weeks apart is designed for preventive immunization, not rapid outbreak response. Ring vaccination only works because protection arrives fast enough to interrupt a transmission chain; a vaccine that takes two months to take effect cannot be measured that way. So its efficacy was never tested in humans. It was licensed instead on immunobridging, inferring human protection from the antibody levels the vaccine produces.
The inference runs through the animals. Researchers measured how antibody levels tracked survival after a lethal dose—higher antibodies, better odds of living. They then measured the antibody levels the vaccine produced in people and asked what survival the animal model would predict for those levels. The result is a predicted survival probability for humans, built entirely on the animal relationship: a mean of 47.8% (95% CI 24.1–66.3%).
That number has to be read against the deliberately harsh test that produced it. The challenge is a high-dose intramuscular infection, at least 200 times the lethal dose, that kills 100% of unvaccinated controls and progresses far faster than human disease, with animals moribund within about a week. The authors are explicit about what this means: because the model is so much harsher than natural infection, the figure “demonstrates that the vaccine is protective in people, rather than being an estimate of human efficacy.” They regard the inferred benefit as conservative. If anything, an underestimate of how well the vaccine would protect in a real outbreak.
It is the only quantitative protection figure that exists for the Janssen regimen, and it was never measured in humans.
How Much Proof Is Enough?
The same epidemic produced two licensed Ebola vaccines by two different standards of evidence.
Ervebo was proven to work in people. The ring trial measured its efficacy directly, under outbreak conditions, with a design built for exactly that purpose. The result, caveats and all, was a human efficacy estimate.
The Janssen regimen was licensed without that. Its two-dose schedule made a ring trial impossible, so its protection was never measured in a person; the only quantitative figure attached to it is a survival probability drawn from an animal model. That leaves a real question: can a model fit on lethally-challenged macaques stand in for an efficacy measurement in people? Regulators decided it could, at least well enough to license the regimen, given that the alternative was no preventive option at all. But deciding the evidence was sufficient to act on is not the same as confirming the vaccine protects people. The bridge was accepted, not verified.
Where does that leave the field for the current outbreak in the DRC and Uganda? The vaccines licensed against Zaire ebolavirus are not approved for Bundibugyo, and whether the rVSV-ZEBOV vaccine offers any cross-protection is genuinely unsettled. One macaque study reported 75% survival against Bundibugyo challenge, but the broader animal record suggests reliable protection across Ebola species likely requires a Bundibugyo-matched antigen or a prime-boost regimen. As Prof Emma Thompson of the MRC–University of Glasgow Centre for Virus Research put it, the non-human-primate work suggests rVSV-ZEBOV “may provide partial heterologous protection against Bundibugyo virus, but this cannot be assumed to translate into reliable protection in people during an outbreak.” Gavi reports that Bundibugyo-specific candidates are being rushed into production: an Oxford/Serum Institute ChAdOx vaccine whose production could be ready in two to three months, and an rVSV-Bundibugyo candidate six to nine months behind. But none yet has efficacy data, and the ChAdOx candidate has no animal or human data at all.
So the field faces the same question again: when there is no time to measure efficacy in people, how much inference from animals is enough to act on? WHO has convened experts to weigh which candidates might enter emergency trials. Gavi chief executive Sania Nishtar has said that roughly 2,000 doses of the licensed Zaire vaccine are already in the DRC, and could be drawn on if a case is made for trialing them against Bundibugyo. Whatever is chosen, the decision will be made on the same axis Ervebo and the Janssen regimen marked out: between protection demonstrated in humans and protection inferred from another species.
Take-Home Messages
The crisis forced a new trial design: ring vaccination — cluster-randomized, immediate-versus-delayed rather than vaccine-versus-placebo, and adaptive. It made efficacy measurable in a waning epidemic without randomizing anyone to no vaccine at all.
The two Zaire vaccines were licensed by two different standards of evidence. Ervebo’s efficacy was measured in humans; the Janssen regimen’s was inferred from animal challenge via immunobridging (a model-predicted 47.8% survival, never measured in people). Both were licensed; only one was shown to protect a person.
The same question is live again. No vaccine is approved for Bundibugyo, the species behind the 2026 outbreak; whether the Zaire vaccine cross-protects is unsettled, and Bundibugyo-specific candidates have little or no efficacy data. WHO must again decide how much animal-inferred evidence is enough to deploy under outbreak conditions.
Footnotes
Originally developed by the Public Health Agency of Canada, licensed in 2010 to BioProtection Systems (a NewLink Genetics subsidiary), and sublicensed to Merck in November 2014 during the West African epidemic.↩︎
After randomization was stopped, the trial was extended to 19 additional clusters in which everyone received immediate vaccination. Pooling these with the randomized clusters produced the more widely quoted figure of 100% (95% CI 79.3–100.0; p=0.0033), based on 0 cases versus 23 cases across 11 affected clusters in the combined never-vaccinated population (eligible delayed plus eligible-never-vaccinated in immediate clusters). Both figures come from the final report (Henao-Restrepo et al., Lancet 2017; 389:505–518); they are different analyses of the same trial. A third figure — 100% (95% CI 74.7–100.0; p=0.0036) — comes from the interim report (Henao-Restrepo et al., Lancet 2015; 386:857–866) and is sometimes conflated with these.↩︎
Built on Janssen’s AdVac vector and Bavarian Nordic’s MVA platform, developed through the EBOVAC consortia under the EU Innovative Medicines Initiative. The European Commission granted marketing authorization under exceptional circumstances on 1 July 2020, following a CHMP positive opinion in May. The EU authorization was subsequently withdrawn on 1 May 2026 at the marketing-authorization holder’s request for commercial reasons, having never been marketed in the EU, though WHO prequalification of the regimen remained in place.↩︎